Clinical trial monitoring is 30-40% of your project costs. At least half of that cost is manual work and SDV activities which can be eliminated with use of modern technologies like electronic source documents and remote risk-based monitoring.
In this post, we take a look at how you can automate routine data management activities and eliminate costly manual labor of your study monitors. In order to eliminate costly people and paper processing, we need to implement a strategy of “management by exception” or as the FDA calls it “remote risk-based monitoring“.
For the benefit of connected medical device developers who are new to clinical trial operations and data management, or new to electronic data capture (EDC) for clinical studies; this post starts with an introduction to FDA guidance on remote risk-based monitoring and goes on to provide some practical suggestions for using your EDC system within a remote risk-based monitoring plan for your connected medical device study.
21 CRF Part 11
Before taking a look at the FDA’s guidance for RBM (remote risk-based monitoring), we note that failure to comply with 21 CFR Part 11 can result in CROs and sponsors being sent citation letters from the FDA, or, far more concerning, a fine of up to $250,000 per each violation.
21 CFR Part 11 requires that you implement controls, including audits, system validations, audit trails, electronic signatures, and documentation for software and systems involved in processing data in your clinical trial. 21 CFR also applies to submissions made to the FDA in electronic format.
Why does the FDA insist on compliance to such a degree? The electronic systems need to be validated as per FDA compliance, first of all, to reduce risk of patient safety
Risk during electronic system operation is categorized by three levels of concern as per the FDA:
1. Major – operation can result in serious injury or death of a subject
2. Moderate – operation can result in a non-serious injury to the subject
3. Minor – operation seems perfectly validated for use and no subject injury is expected
21 CFR Part 11 was written to ensure that electronic data capture and monitoring is just as reliable as paper, when it comes to electronic signatures and trial record keeping and security. Being that an EDC system features time-saving automation abilities for authorization monitoring, it is essential that each system is validated to comply with the FDA regulations.
Risk-based monitoring: quality data and patient compliance
FDA guidance on RBM starts with the notion of identifying a-priori (before the study) critical data and processes, and then performing a risk assessment:
Monitoring activities should focus on preventing or mitigating important and likely sources of error in the conduct, collection, and reporting of critical data and processes necessary for human subject protection and trial integrity. Sponsors should prospectively identify critical data and processes, then perform a risk assessment to identify and understand the risks that could affect the collection of critical data or the performance of critical processes, and then develop a monitoring plan that focuses on the important and likely risks to critical data and processes.
The goals of FDA guidance on RBM are aligned with GCP guidance – namely to assure that high-quality data is being collected, assure high levels of patient compliance to the protocol and closely monitor patient safety in terms of AE (adverse events) and SAE (severe adverse events). during your clinical trial,
In this respect, it is clear that using an EDC is a pre-requisite for RBM.
The key advantage of implementing an EDC system for your clinical studies is the enhanced speed and efficiency that EDC provides over paper data capture and the ability to use the data to automate routine data management and remote monitoring activities.
EDC systems are data capture tools and serve as a data source for RBM which requires looking at aggregates and automating monitoring for deviations,
FDA encourages sponsors to develop monitoring plans that manage important risks to human subjects and data quality and address the challenges of oversight in part by taking advantage of the (technology) innovations in modern clinical trials
With robust RBM integrated into your EDC, you capture high quality data, assure patient compliance to the protocol and reduce the time you spend on data-cleaning at the end of the study, thereby reducing the time you need to produce your statistical report and regulatory submission.
Designing RBM measures
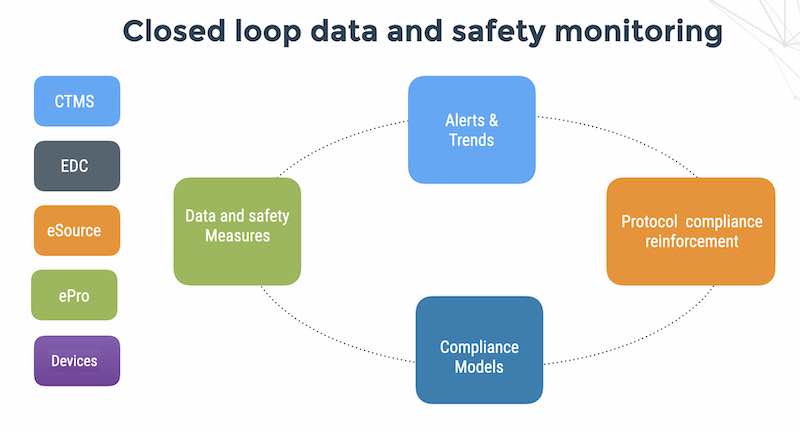
RBM planning takes place during the EDC design stage, when the software itself is being prepared for the planned study. RBM includes a combination of human intelligence, experience with previous similar trials, realistic management expectations, algorithms and metrics designed to track data behavior, notifications for when data is behaving unexpectedly, and, in the case of multiple study sites, real-time alerts for patient-compliance deviations.
The goal of designing RBM is twofold regarding resource allocation and operational efficiency. Instead of evenly assigning resources (clinicians, software specialists, funds, etc.) across study sites, 1.) resources can be saved from wasting time at low-risk sites by tracking data and 2.) assigning risk monitors where they are most needed and put to use.
Data is a prerequisite for decentralization
As we mentioned earlier, Cloud EDC is the first pre-requisite for RBM in terms of its ability to provide a central database and secure decentralized access for monitoring the study conduct and clinical operations of sites and subjects. In addition – you may be collecting data in real-time from patients using ePRO (electronic patient reported outcome) applications and from connected medical devices using the Flask Data API.
Many CROs and sponsors that outsource data monitors take advantage of decentralization. With robust RBM in place, the remote RBM system is able to send alerts in real-time to site coordinators across multiple study sites, who can then respond quickly to patient compliance deviations.
Remote RBM can also help with improvement of the EDC edit checks to better fit with the trials protocol and guarantee better quality data entry. The 21 CFR Part 11 audit trail helps identify who made what mistakes, and who made what corrections and when the initial data entry and administrative editing was performed by the data manager.
Risky studies have noisy data and non-compliant patients
The sooner risks are identified, the sooner they can be mitigated. Real-time, remote data monitoring enables study monitors to save travel time and improve response time to detecting and resolving patient compliance protocol deviations.
When cloud EDC systems are used, managers and monitors can access the system via smartphone, tablet, or any other mobile device that has an internet connection. No matter the time of day, as soon as a risk is flagged by the system it can be addressed and solved. Try doing that with a paper-based system!
Modern data technologies. Old-school customer support.
Use of modern data collection, automated monitoring and decentralization is key to the success of global multi-center studies and to the emerging category of “site-less” studies.
When evaluating an EDC vendor consider you will want to consider the following requirements:
1. Is your study build 21 CFR part 11 compliant?
The software itself may be “designed to be 21 CFR Part 11 compliant” but as a matter of fact, your specific EDC study build needs to be validated. This rules out DIY implementations in general unless you are prepared to pay the cost of a third-party verification and validation service provider.
2. Is real-time, remote risk-based monitoring system a separate module?
Ask. Perhaps the clinical platform technology vendor wants you to buy into an expensive package of additional software and professional services which can add another 6 figures to your costs and another 3-4 months to your implementation.
3. Are you being offered a free lunch?
Maybe. Does your CRO offer a bundle of software products and professional services? How much is included in the CRO functional service package and how much is charged as additional services? Beware of a common practice among CRO, data management and statistics providers to low-ball the EDC and make it back on extras and additional professional services.
4. What kind of technical support do you have for connected medical devices?
Kick the tires first. Does the EDC vendor or partner support a secure REST-based device API out-of-the-box like FlaskData.io API ? Or do you have to consider unwieldy and insecure methods of data extract and import of your device logs?