Question
Are you still doing 100% SDV in your medical device clinical trial?
Here are some facts from medical device clinical trials:
30% of your study budget is for monitoring and 50% of monitoring is for SDV
For a $1M study – you are spending $150k for SDV.
What do you get for your $150K?
Check the EDC and see what percent of the data was updated due to SDV activity. A study we did with 20 medical device studies shows that it is less than 1/3 of 1% for connected medical devices using ePRO ( the endpoint/patient compliance data is collected electronically).
Your return on investment was $525.
What does Risk-Based Monitoring mean for CROs?
It basically means a golden opportunity to shake-down the customer.
The goal of any medical device clinical trial is to test the efficacy and safety of innovative pharmaceutical products and medical devices that improve quality of life, cure disease and protect patients.
The goal of any CRO is to wring as much money as possible from their customers with smoke and mirrors.
Contract research organizations (CROs) should approach risk-based monitoring (RBM) as a top priority for clinical studies. Use of modern data technologies for eSource and remote risk-based monitoring will reduce non-value added rework, people and paper in medical device clinical trials and speed up time to statistical report.
I’m not suggesting that CROs stop doing their job. I’m suggesting that CROs focus on their main value-add – complying with GCP for patient safety, data quality and protocol compliance.
Using a combination of a risk-based approach and modern ePRO and eSource technology assures that data collected is reliable, monitor patient safety issues and assure data quality risk in studies.
In this post, we are going to go over how remote risk-based monitoring can help your current study and also provide value for future drug and medical device development.
What is risk-based monitoring?
Risk-based monitoring entails the mitigation of risks during medical device clinical trial that a CRO is conducting through the process of identifying, assessing, and then monitoring the risks associated with patient safety throughout the course of the study.
In August 2013 the FDA issued draft guidance for “A Risk-based approach to monitoring”
“The overarching goal of this guidance is to enhance human subject protection and the quality of clinical trial data by focusing sponsor oversight on the most important aspects of study conduct and reporting”
The draft guidance includes three steps in a risk-based approach to monitoring:
1. Identifying critical data and processes.
2. To accurately monitor the quality of a study and the safety of its patients, sponsors must know which elements are vital for each particular study, including informed consent to eligibility screening and tracking of adverse events.
3. Performing an assessment of risk. Risk assessments requires determining specific causes of risk and the effect of study errors pertaining to risk.
4. Developing a comprehensive monitoring plan. According to the FDA’s guidance regulations, RBM plans should “describe the monitoring methods, responsibilities, and requirements of the trial.” Planning is responsible for communicating risks and monitoring procedures to each party involved in trial RBM.
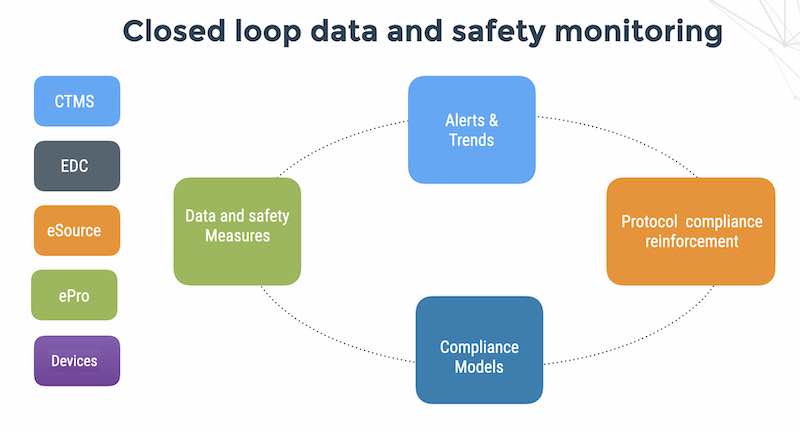
With Case Report Forms (CRFs) now being completed with cloud electronic data capture (EDC) software systems, (or entered directly using eSD (electronic source documents) or ePro (electronic patient reported outcomes), it is possible to to collect CRF data into a centralized database, accessible by all parties, along with a full register of operational, quality controls, efficacy and clinical safety data from all sites, subjects and devices across multiple studies.
Remote risk-based monitoring algorithms are designed for scanning the data for specific and calculate specific metrics that trigger alerts to the clinical data management team – for example excessive query rates or the cycle time from patient visit complete to eCRF data entered both of which are indicative of data quality issues at sites
Remote risk-based monitoring metrics may be considered in two main categories: patient safety risk (for example trends of vital signs) and data quality risk (for example long data entry cycle times).
When human study monitors visit sites once every 4-6 weeks, it is hard to catch slow-moving, high risk events. Human brains are not good at identifying and classifying small numbers of slow-moving events, although they can easily identify a face moving quickly across a basketball court. Computer algorithms are outstanding at identifying anomalies in time-based data found in medical device clinical trials – and this is where RBM will have its biggest payback for you in your study.
What comes next?
What comes next is to make an accurate assessment of the data model. A single number, or a graph displaying data points, data in any form, has no meaning until the data is coherent and understood by a clinician with how the data is understood, and interpreted with how it can produce increased or mitigated risk to the patient, and the study as a whole.
Risk based monitoring and transparency
Take this for example: if a study has a greater than expected negative-result reporting rate, are we talking about the great attention to detail of the study staff, or the negative performance of the study drug? How to determine this?
The answer is that you need to dive in deeper, and have monitors in place that are not only looking at the data, but observing whether the site is performing at the maximum study level, or whether the data is plainly reflecting that the drug is just wrong for the study subjects.
An essential component in performing RBM is that it is required to guarantee that it can ensure that your data collected either indicates that risk is assessed, and that analysts can repeat, from site-to-site (if multiple sites are used) consistent performance that will protect study subjects, and reduce the time and effort it takes to identify risks.
The next crucial course of action during the study is to use the findings to take a direct course of action. With a clear understanding of a site’s specific risk level, CROs should be designated to visit the site(s) and take appropriate measures to alleviate site risks.
With a coherent and thorough understanding of study risks, CROs will take the necessary steps to not only reduce patient risks, but reduce future patient risks, helping your studies run more efficiently and save future costs.
What to look out for, and how to approach challenges
In our experience it has been proven that combined analysis, EDC system automation, visualisation tools, along with the monitors’ innate abilities, established processes for corrective actions have resulted in an efficient and effective uncovering of risks. This can, and will, put data and patients, and thus the entire study in jeopardy. What CRO monitors should look out for include:
1. Fabricated, false data
2. Data omissions
3. Deviations from protocol, poorly trained clinician data submissions
4. Patient compliance to the protocol
Take for example if there was data that showed an alarmingly high rate of a rise in blood pressure for study subjects. Monitors should not only be looking into the increase in blood pressure, but also examining whether the data being submitted is accurate. Monitors should think about whether the submitted data is wrong, or if the data is sound and the drug is at fault.
Fostering a culture of integrity and transparency for study success
A risk-based approach and modern eSource and ePRO should be your culture of study operations. The goal is to ensure that a CRO is approaching studies with not only patient care in mind, but also study efficiency and lower study costs. This is based upon past assessment of data monitoring and monitoring abilities. This is the future for patient compliance, data quality assurance and patient safety.