CLINICAL TRIAL READINESS ASSESSMENT
Are you really ready to run a clinical trial? How can you best assess your clinical trial readiness? We work with C-level executives and management

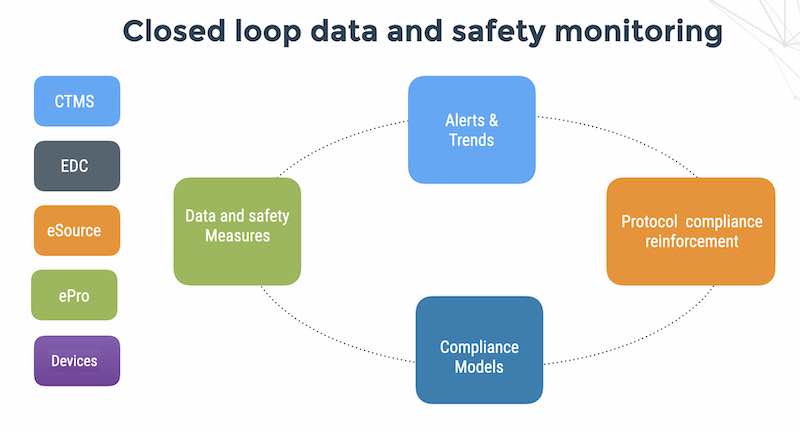
Assuring protocol compliance without checklists
At a site monitoring visit, the CRA works on assuring protocol compliance. Assuring protocol compliance is one of the 3 pillars of GCP: The 3

Home alone and in a clinical trial
1 in 7 American adults live alone COVID and social isolation is part of our lives. During COVID, it became easier to recruit patients for

Preventing drug counterfeiting
Counterfeiting is old as money itself. Drug counterfeiting is no exception. In this post, we’ll share an analytical approach that you can use to estimate
What risks really count for your clinical trials?
Is there a “black-box” risk management solution for your clinical trial? What clinical trial risks are the most important for you and your company? Risk

Dealing with information junkies in decentralized clinical trials
DecentraIized clinical trials software vendor Medable is doing an outstanding job in understanding and executing an online work-flow between sites and patients at home.. This understanding and

Preventing patient data leaks
6 ways to protect patient data in your eClinical and digital health applications Patient data leaks is much more than patient privacy. Patient data leaks

How to ensure patient adherence in decentralized clinical trials
Patient adherence in clinical trials without technology How can you assure patient adherence to the protocol, when you don’t see the patient? Life science companies

How to meet the 10 top challenges in Phase 1 clinical trials
Phase 1 challenges are unlike larger Phase 2, Phase 3 studies. The science is still unsure. The the clinical operations team at a startup may
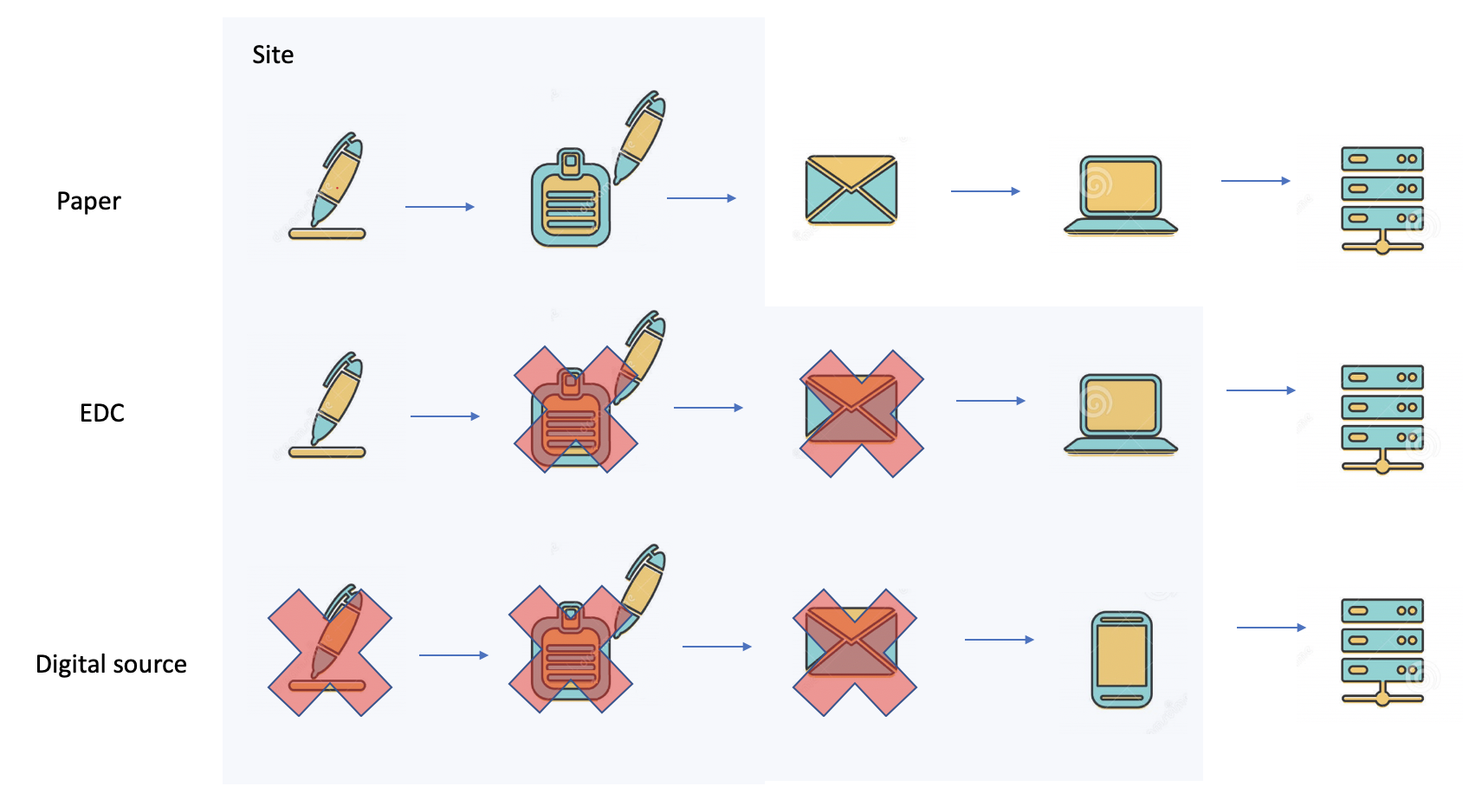
SDV sucks
How professional services delay cures for patients. Why SDV Sucks In the early 50s of the previous century, nobody knew that 70 years later clinical
Clinical data management, payment for outcomes
Payment for outcomes using GCP metrics is a way to significantly reduce clinical operations costs and increase value-for-money from your CRO or DCT vendor. Payment

GCP for clinical trials – patterns of low-concern and high-impact
How to assure GCP for clinical trials in the best way? In this post, we will show you how to assure good clinical practice in

Why you do not want to unify data in your clinical trial
Unified clinical data sets – good or bad? Unification of data from patient medical records, hospital reports and clinical trial protocols is a tempting yet

Knowledge Prostitution in clinical data
Friday, a client asked me about privacy, data security and social networking in clinical trials. The client told me that they were considering using a

Career development for clinical data managers
A good clinical data manager is an essential piece of running a clinical trial. A well-trained and responsibility clinical data manager is a an important

Did you wait until the last minute to choose vendors for your clinical trial?
Your dream outcome. Helping cancer patients. Doing it right. Recruiting quickly and efficiently and getting a diverse population into your study. You have noble intentions

Data capture by sites is activity, not achievement
Never mistake data collection activity for an achievement Recruiting and caring for patients (whether at home or on site) is a research site responsibility. Capturing
Patient compliance in clinical trials – the elephant in the room
Clinical trials cost a fortune. When a patient is non-compliant during the run-in/washout period, he/she is dropped out of the study. This costs the sponsor

Living in an ideal world where the site coordinator is not overwhelmed by IT
Tigran examines the idea of using EDC edit checks to assure patient compliance to the protocol. How should I assure patient compliance to the protocol in
Are you neglecting security incident response in your DCT?
Let me ask you 3 questions. If you answer Yes to all 3 – read this post, if not, then move on. Do you assume

The 20M unmet needs of clinical trials according to social media and Google
If you google unmet needs in clinical trials in clinical trials – you get 20M hits. If you google “unmet needs in clinical trials” (for

The last mile in clinical trials
First mover advantage for clinical trials Although the notion of same day delivery seems to be recent, in fact, competitive advantage always went to those
How understanding culture reduces risk in your clinical trials
It’s during the war the Russians are waging on Ukraine and I got on a thread on a blog about why Putin is so violent.

3 ways that assure protocol compliance and data integrity in your clinical trials
In this short essay, I’ll take a closer look at how the clinical trial supply chain is evolving. There is now rapid change from traditional

HIPAA compliance for your clinical trials
The golden rules of HIPAA compliance for your clinical trials Flask Data provides a one-stop cloud subscription for EDC, data management and statistics. Welcome to the

Our culture – diverse, committed and engaged with customers
Courtesy of Yaron Koler. It’s not about the EDC system. It’s about our values. There are 2 reasons for this: The first reason, is that

The effectiveness of access controls in clinical data exchange
Sharepoint or not. Hint – not. Transferring a dump of clinical trial data? It was recently suggested to us by a data manager, that he
If you see it in a dashboard, its too late
Thoughts on monitoring patient data and safety in clinical trials The future: tech-driven data and safety monitoring Running clinical trials with the help of tech
Github – A community of practice for the clinical trial community
Encouraging a participatory culture in clinical trials Founder and CEO of flaskdata.io, Danny Lieberman wonders if the phenomenal success of the participatory code culture on GitHub
The roles of trust, security and privacy in clinical trials
Trust, security and privacy is a cornerstone in clinical trials pri·va·cy/ˈprīvəsē/ The state or condition of being free from being observed or disturbed by other people.
Use ML and AI to automate clinical trial operations
I’m always curious to learn new ideas from other disciplines and apply them for my own selfish purposes. In this case – the AIOps model for
High-performance clinical teams speak softly
The advantages of speaking softly I started thinking about the constraints on eClinical technology for decentralized clinical trials and patient-centric product development. The best planned
How to get digital health apps to work together – the power of simplicity
Sunrise off the coast of the Dead Sea in Israel We are using too many buzzwords to defeat SARS-COV-2 I thought about buzz-words last night.
The LA Freeway model of clinical monitoring
A freeway paradigm helps explain why onsite visits by study monitors don’t work and helps us plan and implement an effective system for protocol compliance monitoring of all sites,
How to swim in the cold water of decentralized clinical trials
(The water in the pool in Cascais in October was < 12 degrees) There are over 135 COVID-19 vaccine candidates in the pipeline at the
3 things to do before you spend money on a HIPAA consultant for your clinical trial
Flaskdata specializes in same data data and safety solutions for clinical trials. Flaskdata is a technology company specializing in clinical datamanagement and monitoring. We are accomplished
Hack back the user interface for clinical trials
As part of my campaign for site-coordinator and study-monitor centric clinical trials; we first need to understand how to exploit a vulnerability in human psychology.
A better tomorrow for clinical trials
A better tomorrow – Times of crisis usher in new mindsets By David Laxer. Spoken from the heart. In these trying days, as we adjust to
7 tips for an agile healthtech startup
It’s a time when we are all remote-workers. Startups looking for new ways to add value to customers. Large pharmas looking for ways to innovate
Streaming clinical trials in a post-Corona future
Last week, I wrote about using automated detection and response technology to mitigate the next Corona pandemic. Today – we’ll take a closer look at
So what’s wrong with 1990s EDC systems?
Make no doubt about it, the EDC systems of 2020 are using a 1990’s design. (OK – granted, there are some innovators out there like
I love being a CRA, but the role as it exists today is obsolete.
I think that COVID-19 will be the death knell for on-site monitoring visits and SDV. Predictions for 2020 and the next generation of clinical
Using automated detection and response technology mitigate the next Corona pandemic
What happens the day after? What happens next winter? Sure – we must find effective treatment and vaccines. Sure – we need to reduce or
10 ways to detect people who are a threat to your clinical trial
Flaskdata.io helps Life Science CxO teams outcompete using continuous data feeds from patients, devices and investigators mixed with a slice of patient compliance automation. One
Competitive buzzwords in EDC companies
We recently did a presentation to a person at one of the big 4 pharma. His job title was Senior IT Project Manager Specialized in
Develop project management competencies to speed up your clinical trials
The biggest barrier to shortening clinical trial data cycle times is not recruitment. It is not having a fancy UI for self-service eCRF forms design.
5 ways to make your clinical trials run real fast
This week, we had a few charming examples of risk management in clinical trials with several of our customers. I started thinking about what we
Temperature excursions and APIs to reduce study monitor work
I did a lot of local excursions the past 3 days – Jerusalem, Tel Aviv, Herzliya and Haifa. For some reason, the conversations with 2
Doctor-Patient Communication – the key to success and the struggle to succeed.
Katherine Murphy, Chief Executive of the Patients Association London once said, “The huge rise in complaints in relation to communication and a lack of respect
Urban medical legends
Because I was trained as a solid-state physicist I am skeptical of many medical claims – including the efficacy of digital health apps. Gina Kolata
What takes precedence? GCP or hospital network security?
This is a piece I wrote a while back on my medical device security blog – Cybersecurity for medical devices. One of the biggest challenge
Why Microsoft is evil for medical devices
Another hot day in paradise. Sunny and 34C. Not a disaster but still a PITA We just spent 2 days bug-fixing and regression-testing code that
How to measure clinical response in medical device clinical trials
It is 19:15 and daylight savings time. It is too hot to go out and run or bike. Time to write. Today we were helping
Bionic M2M: Are Skin-mounted M2M devices – the future of clinical trials?
There is a lot of hype about wearables. One of the best ways to correlate patient compliance with patient biometrics is for the patient to
The gap between the proletariat and Medidata (or should I say Dassault)
We need a better UX before [TLA] integration The sheer number and variety of eClinical software companies and buzzwords confuses me. There is EDC, CTMS,
Obsessed with patient compliance
Obsessed with patient compliance I’m watching a series of short videos done by Techstars founder Brad Feld. Brad talks about founders needing to be obsessive. I
How to annoy your eClinical platform vendor
Every question is a cry to understand the world. There is no such thing as a dumb question. Carl Sagan In this guest post, my
Good strategy bad strategy for study monitors in connected device studies
Friday is an off-day in Israel and I try to work on projects or read. I am now reading Richard Rumelt’s book Good strategy Bad
How to improve patient compliance in your medical device study
Here’s an idea that will make you slap your forehead. You can just stop transcribing case reports on paper. FDA eSource guidance recommends direct data entry
Treating EDC Induced Dissociative Panic Disorder
There is considerable online discussion about real-world data in clinical trials, virtual trials, digital trials, medical IoT, wearables, AI, machine learning for finding best candidates
Killed by code in your connected medical device
Are we more concerned with politicians with pacemakers or families with large numbers of connected medical devices? Back in 2011, I thought it would only be
Improving patient compliance to medical device protocols with threat models
To paraphrase Lord Kelvin – “You cannot improve what you cannot measure”. I have about 10′ before Shabbat and I wanted to offer 2 possible
4 strategies to get connected medical devices faster to FDA submission
Introduction Better designs, site-less trials, all-digital data collection and PCM (patient compliance monitoring) can all save time and money in connected medical device clinical trials.
How to become an insights-driven clinical operations manager
In my post Putting lipstick on a pig of eCRF I noted that good online systems do not use paper paradigms. In this post –
The golden rule for digital therapeutics and connected medical devices
He who has the gold rules. That’s all you need to know when it comes to privacy compliance. In the past 5 years, a lot
Putting lipstick on the pig of electronic CRF?
Good online systems do not use paper paradigms. In this post – I will try and entertain you with historical perspective and quantitative tools for
Israel Biomed 2019-the high-social, low stress STEM conference
Impressions from Biomed 2019 in Tel Aviv This week was the annual 3 day Biomed/MIXiii (I have no idea what MIXiii means btw) conference in
Living off generic solutions developed in the past
I recently read some posts on Fred Wilson’s blog and it was impressive that he writes every day. I’ve fallen into the trap of collecting
Perverse incentives
The perverse incentive for the high costs of medical devices and delay to market The CRO outsourcing model and high US hospital prices result in
Patient compliance – the billion dollar question
The high failure rate of drug trials The high failure rate of drugs in clinical trials, especially in the later stages of development, is a
Killed by code – back to the future
I hope that the code in your digital therapeutic for treating autistic children, doesn’t look like this. Back in 2011, I thought it would only be
Teetering on the precipice of medical device/digital health clinical trials
Danny teeters on the edge of the precipice of privacy and security. Step on the brakes not on the gas and don’t look down. Take
When is patient compliance important in medical device clinical trials?
In this post, Danny Lieberman, founder of flaskdata.io , discusses when patient compliance is crucial for your medical device clinical trial and when patient compliance is
Israeli Medical device innovation for high patient compliance
One of the most challenging problems in medical device clinical trials and in real-life is how to achieve high levels of patient compliance to the
Invisible gorillas and detection of adverse events in medical device trials
What is easier to detect in your study – Slow-moving or fast moving deviations? This post considers human frailty and strengths. We recently performed a
Why paper is not an option for your medical device clinical trial
This is a piece David wrote a couple of years ago originally entitled “Why you cannot afford to use paper in your first Phase I
A structured 7 step process for risk assessment of a decentralized clinical trial
In this essay, I discuss a systematic methodology for evaluating risk in your decentralized clinical trial. This is a methodology that has proven itself in
Why EDC is essential for any medical device clinical trial
This is a post David wrote a while back and it still seems relevant. If you would have asked me 2 years ago – I
What does risk-based monitoring mean for CROS?
Contract research organizations (CROs) should implement risk-based monitoring (RBM) as a top priority for medical device clinical studies. Use of modern data technologies for remote
How to sustain high patient compliance in medical device trials
A comparison between pharma trials and medical device clinical trials The differences between medical device trials and drug efficacy studies are similar to the differences
Is social networking a threat or an opportunity for patient compliance?
How social networking helps medical device clinical trials achieve high rates of recruitment and patient compliance.
The role of your biostatistician for success in medical device clinical trials
The importance of biostatistics in medical device clinical trials In order to understand the importance of biostatistics in medical device clinical trials we talked to
Strong patient adherence in real life starts with strong people management
Patient adherence in real-life starts in clinical trials determining the safety, side effects and efficacy of the intervention, whether a drug or a medical device.
What real-time data and Risk-based monitoring mean for your CRO
A widely neglected factor in cost-effective risk-based clinical trial monitoring is availability and accessibility of data. RBM methods used by a central clinical trial monitoring
The 3 tenets for designing a clinical data management system
Abstract: This post reviews the importance of 1) proper study design, 2) good data modeling and 3) realistic estimation of project timetables. The article concludes
Medical device clinical trials – not for the faint of heart
Patients in medical device clinical trials are on their phones. On their phones for WhatsApp and for monitoring chronic conditions and reporting outcomes at home, at work
Millennials are the future of clinical trial data management
Millennials, born between 1980 and 2000 and the first native generation of the digital age, are the quickly approaching additions to the modern workforce. Regardless
Risk based monitoring. It’s about the people.
It will come as no surprise to anyone who is actively involved in clinical trial operations that the heart of the matter in clinical trial monitoring is
Important EDC features for medical device clinical trials
Medidata Rave and its CTMS companion product iMedidata are a far more comprehensive solution than OpenClinica but when you choose EDC software for medical device
Implementing an EDC to take advantage of risk-based monitoring
Clinical trial monitoring is 30-40% of your project costs. At least half of that cost is manual work and SDV activities which can be
Integrating mHealth in medical device clinical trials
Introduction mHealth evolved largely an application for developing countries, due to the rapid growth of mobile phone penetration in low-income nations. But mHealth is now
How to secure your data in mobile medical device clinical trials
So you are getting ready to run medical device clinical trials with your mobile medical app or a medical appliance that is connected to the
Homeostasis and medical device clinical trials
Danny talks about how to strike a good balance between people and technology for monitoring medical device clinical trials. Are real-time alerts too much of
The great ripoff of SDV in medical device studies
Question Are you still doing 100% SDV in your medical device clinical trial? Here are some facts from medical device clinical trials: 30% of your
People, not digital for supporting patients in medical device clinical trials
1 in 7 American adults live alone Medical device clinical trials are performed under conditions that are near to real-life use by the patient. That
Why medical device studies need business controls
There are some interesting analogies between cyber security and medical device clinical trials from a risk management perspective. Both areas are complex, vulnerable to human
Why healthcare IoT devices need data monitoring
Use of healthcare IoT devices for sensing patient vital signs enables fast and cost-effective remote monitoring of patient safety and data quality – however the
The 2 most common mistakes in Clinical Research Data Management
Another survey piece that David wrote about common mistakes in clinical data management and some basic controls to stay from the common issues. Use automated
The best alternative to paper in medical device clinical trials
There is an urban legend that paper is cheaper than EDC $1000/subject for paper-based data management (the going rate in Israel) is a lucrative business
Why medical IoT is outside your comfort zone
Connected medical devices or (medical IoT – Internet of Things) outnumber people. This is an article David wrote about 1.5 years ago. It is a
Using AI to assure patient compliance in heart failure patients
Can AI be used to help patients with heart failure? Each year cardiovascular disease (CVD) causes 3.9 million deaths in Europe and over 1.8 million
why a medical device is now the biggest risk in your clinical trial
Of all of the connections brought about by the Internet of Things, nothing is more frightening than the notion of an unsecured medical device. The
A word to Teva on firing employees and assuring data security
To be able to do something before it exists, sense before it becomes active, and see before it sprouts. The Book of Balance and Harmony
The pay-off for reducing cycle time in medical device clinical trials
Well – it certainly isn’t science and technology innovation or even the FDA.At a recent executive roundtable, according to Tufts University’s Center for the Study of
How to overcome 5 eSource implementation challenges
Jenya wrote a piece about the challenges of clinical trials operations change management for regulatory people who have to work with medical technology developers and I
Cost effective EDC for medical device clinical trials
There is a saying in American English dating back to the 1940’s – “Call me when you have a nickel in your pocket”. With limited
The key is not first to eSource, the key is smart to market
This post is not for the Pfizers, Novartis, Merck and GSK giants of the life science industry. Its for the innovators, the smaller, creative life science companies
Assuring patient compliance to the study protocol-spending smart on monitoring
Today I want to go beyond having compelling ideas, a team and a great market opportunity and talk about what you need to successfully execute
Dates: the silent death in medical device clinical trials
Jenya Konikov-Rozenman Jenya is a co-founder and VP Clinical at Flaskdata.io. Jenya has a masters degree in biotechnology from the Hebrew University and is a
An attack modeling approach to medical device clinical trials
What does taking off your shoes and belt in the airport have in common with risk assessment in clinical trials? Today we talk about the drawbacks of
Why paper should be your first choice for a medical device clinical trial
6 reasons to use paper in your clinical trial You’ve been considering cloud EDC for your next clinical trial, but you are put off by
5 ways to reduce costs and accelerate medical device clinical trials
Time is money The economic model of a multi-center clinical trial is based on a commercial biotech/biomed/pharma company sponsoring and funding clinical research in order
5 critical factors in choosing software for clinical data management
Here are some practical guidelines to selecting an EDC system. Some basic blocking and tackling. Always good. EDC not paper Electronic data capture (EDC) systems
5 key questions for choosing clinical data management software
Pricing from the big EDC vendors is complex and confusing and the overlap between CRO clinical data management services and their EDC solutions locks you
3 critical success factors for patient compliance automation
Even if you are new in pharmaceutical or medical device clinical trial industry, it is more probable than not that you recognise the value of
3 practical tips for cost-effective clinical monitoring
Originally posted by Jenya Konikov-Rozenman on May 31, 2016 in Clinical Trial Monitoring Can you use technology to reduce the costs of your clinical trial and
WannaCrypt attacks
For your IMMEDIATE notice: If you run medical device Windows management consoles, run Windows Update and update your machine NOW. This is my professional advice
What is more important – patient safety or hospital IT?
What is more important – patient safety or the health of the enterprise hospital Windows network? What is more important – writing secure code or
Why HIPAA Policies and Procedures are not copy and paste
Compliance from Dr. Google is a very bad idea. Searching for HIPAA Security Rule compliance yields about 1.8Million hits on Google. Some of the information is outdated
Encryption and medical device cyber security
I have written pieces here, here, here and here on why encryption should be a required security countermeasure for network medical devices – but curiously, the HIPAA
Procedures are not a substitute for ethical behavior
Are procedures a substitute for responsible and ethical behavior? The behavior of former secretary of State (and Presidential race loser) Hilary Clinton is an important
The chasm between FDA regulatory and cyber security
When a Risk Analysis is not a Risk analysis Superficially at least, there is not a lot of difference between a threat analysis that
PCI DSS is a standard for the card associations not for your business
I recently saw a post from a blog on a corporate web site from a company called Cloud compliance, entitled “Compliance is the New
Why the Clinton data leaks matter
In the middle of a US Presidential election that will certainly become more contrast-focused (as politically correct Americans like to call mud-slinging), the Clinton data
Why audit and risk management do not mitigate risk – part II
In my previous post Risk does not walk alone – I noted both the importance and often ignored lack of relevance of internal audit and corporate
Risk does not walk alone
Israeli biomed companies often ask us about the roles of audit and risk management in their HIPAA security and compliance activities. At the eHealth conference
The 2 big data disconnects in clinical data management
Why data is important There are 2 reasons: Determining the results of your clinical trials depends on the data Governance of the clinical trial depends on
Connecting with serious ill children at the Jordan River Village
Working in a startup like FlaskData.io developing cloud-based clinical data management software is like a triathlon ( I did the Eilat triathlon in 2013 so
2 mistakes you do not want to make in your medical device clinical trial
Jenya Konikov-Rozenman Jenya is a co-founder and VP Clinical at Flaskdata.io. Jenya has a masters degree in biotechnology from the Hebrew University and is a
How do you know that your personal health data is secure in the cloud?
Modern system architecture for medical devices is a triangle of Medical device, Mobile app and Cloud services (storing, processing and visualizing health data collected from the device). This
3 things a medical device vendor must do for security incident response
You are VP R&D or CEO or regulatory and compliance officer at a medical device company. Your medical devices measure something (blood sugar, urine analysis, facial anomalies,
Refreshing your HIPAA Security Rule compliance
Clients frequently ask us questions like this. Danny, I have a quick question about our HIPAA compliance that we achieved back in early 2013. Since then we have released a
Forging partnerships for high patient compliance
Clarifying ownership of patient compliance in medical device clinical trials requires building risk management into the operation and forging a good partnership between clinical operations,
Serious fun at the Jordan River Village
Working in a startup like FlaskData.io is a combination of riding a roller-coaster and doing a 100K bike ride. There are downs (losing deals) and ups (winning deals),
Invisible gorillas and clinical trial monitoring
Danny Lieberman Invisible gorillas and detection of severe adverse events. This weekly episode considers human frailty and strengths. We recently performed a retrospective study of
Airport security versus clinical trial monitoring
Airport security versus clinical trial risk management What does taking off your shoes and belt in the airport have in common with risk assessment in
Privacy, Security, HIPAA and you.
Medical devices, mobile apps, Web applications – storing data in the cloud, sharing with hospitals and doctors. How do I comply with HIPAA? What applies
Why the CRO model for drug and device trials is broken and how to fix it
Who said: ‘If you are not part of the solution, you must be part of the problem’? This appears to be a misquotation of Eldridge Cleaver
5 things that make mHealth apps successful
In case you were thinking about scanning this post before scooting over to sign up to get 30 days free on our new managed Cloud clinical
What makes mHealth apps flavorgasmic?
When eating food so good that you let out an involuntary moan, usually the first bite; also as an adjective – flavorgasmic. November 30, 2008 Urban
Crossing the valley of death of clinical trial monitoring
When hype exceeds adoption As a matter of fact, hype always succeeds adoption and rightly so – because hype is a way of getting our
ICH GCP and EU Regulations impact on clinical trial monitoring
October 2015: EU standards bodies are so far behind the curve that they now emphasize CSV (which been around since 1995 or so and formally documented
You can DIY a chair from IKEA. You cannot DIY your medical device clinical trial
Jenya Konikov-Rozenman Jenya is a co-founder and VP Clinical at Flaskdata.io. Jenya has a masters degree in biotechnology from the Hebrew University and is
5 fast tips to make medical device clinical trials faster and cheaper
1. Keep your design simple. More then 100 parameters per subject is too much. 2. Plan your time. Start 6 months before first patient in.
Why your security is worse than you think
Thoughts for Yom Kippur – the Jewish day of atonement – coming up next Wed. Security on modern operating systems (Windows, OS/X, iOS, Android, Linux)
14 years after 9/11, more connected, more social, more violent
Friday, today is the 14’th anniversary of the Al Queda attack on the US in New York on 9/11/2001. The world today is more connected,
The importance of risk analysis for HIPAA compliance
A chain of risk analysis The HIPAA Final Rule creates a chain of risk analysis and compliance from the hospital, downstream to the business associates who handle
On Shoshin and Software Security
I am an independent software security consultant specializing in medical device security and HIPAA compliance in Israel. I use the state-of-the art PTA –
Dealing with DLP and privacy
Dealing with DLP and privacy It’s a long hot summer here in the Middle East and with 2/3 of the office out on vacation, you have some time
What is PHI?
Software Associates specialize in HIPAA security and compliance for Israeli medical device companies – and 2 questions always come up: “What is PHI?” and “What is electronically protected
10 ways to detect employees who are a threat to PHI
Software Associates specializes in software security and privacy compliance for medical device vendors in Israel. One of the great things about working with Israeli
The top 5 things a medical device vendor should do for HIPAA compliance
We specialize in software security assessments, FDA cyber-security and HIPAA compliance for medical device vendors in Israel. The first question that every medical device vendor CEO
Shock therapy for medical device malware
Israel has over 700 medical device vendors. Sometimes it seems like half of them are attaching to the cloud and the other are developing mobile
The death of the anti-virus
Does anti-virus really protect your data? Additional security controls do not necessarily reduce risk. Installing more security products is never a free lunch and
It’s friends and family breaching patient privacy – not Estonian hackers.
A 2011 HIPAA patient privacy violation in Canada, where an imaging technician accessed the medical records of her ex-husband’s girlfriend is illustrative of unauthorized disclosure
How to share information securely in online support groups
Pathcare is a HIPAA-compliant service for sharing and private messaging with support group members and support group leaders and faciliators. Inside the Pathcare private social network for healthcare– you don’t have
How to protect your personal information from medical data theft
Private, personal information can be bought and sold on the black market for as little as fifty cents to a dollar, according to a report
A new agenda for Israeli education.
הצעה לסדר יום אחר בבית הספר בישראל הזמן – עוד מעט ראש השנה של איזה שנה עברית ששמה לא זכורה למרביתנו. המקום – אי-שם במדינת
Mobile security: Risks of 2014 and beyond
Mobile security: Risks of 2014 and beyond These days the vast majority of us own mobile phones with some of us owning more than one, not
Why Google is a bad idea for security and compliance
Dear consultant, I worry because so many of the best practices documents I read say that we need to store data in the cloud in
Picking Your Way Through the Mime Field
Picking Your Way Through the Mime Field We’re a professional software security consultancy and experienced software developers. Almost 10 years, one of our partners proposed
Kick start your European privacy compliance
The CNIL’s Sanctions Committee issues a 150 000 € monetary penalty to GOOGLE Inc. On 3 January 2014, the CNIL’s Sanctions Committee issued a 150 000
Is Your Small Business Safe From Cyberattacks?
Of the 855 data breaches Verizon examined in its 2012 Data Breach Investigations Study, 71 percent occurred at businesses with fewer than 100 employees. The
Out of control with BYOD in your hospital?
The number of bring your own device (BYOD) workplaces is increasing. Hospitals are certainly no exception with nursing staff, doctors and contractors bringing their own
What is your take on anti-virus in medical devices?
A check-box IT requirement for medical devices on the hospital network is installation of anti-virus software even though most devices don’t have network connectivity and
Why anti-virus doesn’t work for medical devices
Are you checking off medical device security in your hospital with anti-virus: falling for security theater; feeling secure and enjoying the show, but in fact being
How to Save Your Data and Reputation if You Lose Your BlackBerry
5 years ago, an analysis we did of 150 data breach events showed that over 40% of the data breach events were due to stolen
מלחמת סייבר – לתקוף את המרקם החברתי של האקרים ולא להתגונן
הפרדיגמה הצבאית קונבנציונלית אינה מתאימה לאבטחת סייבר מדיניות Cyber Security של מדינות שונות עוצבה בידי הצבא ולכן באופן מסורתיcyber security נתפשת רק בהקשר של אסטרטגיית
4 steps to small business security
Software Associates specializes in security and compliance for biomed. Many of our biomed clients are small 3-10 person startups working out of a small office
Health Information Technology Patient Safety Action & Surveillance Plan
This is a quick update on two new documents released by the HHS and the IMDRF: Health Information Technology Patient Safety Action & Surveillance Plan
Why security defenses are a mistake
Security defenses don’t improve our understanding of the root causes of data breaches Why is this so? Because when you defend against a data breach
Software in Medical Devices – Update
We have previously written about various aspects of the software development process, especially, the verification and validation activities in implanted and invasive medical devices. Here is
The dangers of default passwords – 37% of Data Breaches Found to be Malicious Attacks
A malicious attack by malware or spear phishing on valuable data assets like PHI (protected health information) exploits known vulnerabilities and one of the most
Is cyber security and mobile device management important in the healthcare industry?
Is cyber security and mobile device management important in the healthcare industry? Healthcare and technology go hand in glove more than almost any other sector in today’s
Is your HIPAA security like a washing machine?
Is your HIPAA security management like a washing machine? Most security appliance vendors use fluffy charts with a 4 step “information risk management” cycle. It’s
The facts of life for HIPAA business associates
If you are a biomed vendor and you collect any kind of PHI (protected health information) in your medical device or store information in the
How to use BI to improve healthcare IT security
Information technology management is about executing predictable business processes. Information Security Management is about reducing the impact of unpredictable attacks to your healthcare provider organization.
Snake Oil 2.0 – why more data is bad
Why more data is bad Remember the old joke regarding college degrees? BS = Bull Shit, MS = More Shit and PhD == Piled Higher
Auditing healthcare IT security and privacy with multiple threat scenarios
Is there a way to break out of the security checklist mentality? IT security auditors commonly use standard/fixed checklists, often based on the compliance regulation
The best cybersecurity strategy may be counter-terror
Danny Lieberman suggests that a demand-side strategy with peer-review may work best for cyber-security. A conventional military paradigm does not work for cyber-security Government cyber
Why big data for healthcare is dangerous and wrong
The Mckinsey Global Institute recently published a report entitled – Big data: The next frontier for innovation, competition, and productivity . The Mckinsey Global Institute report
The mistakes you will make on your next cloud project
Are you considering cloud security in the abstract or cloud security in your software? Looking at cloud security issues in the abstract, we see 4
Bionic M2M: Are Skin-mounted M2M devices – the future of eHealth?
In the popular American TV series that aired on ABC in the 70s, Steve Austin is the “Six million Dollar Man”, a former astronaut with
The Private Social Network for healthcare
In his post on the Pathcare blog, I trust you to keep this private, Danny Lieberman talked about the roles that trust, security and privacy play
Can I use Dropbox for storing healthcare data?
First of all, I’m a great fan of Dropbox. It’s easy to use, fast, runs on Windows, Mac and Linux and that means you can
How to keep secrets in healthcare online
The roles of trust, security and privacy in healthcare. If President Obama had told his psychiatrist he was gay, you can bet that it would
Five things a healthcare CIO can do to improve security
A metaphor I like to use with clients compares security vulnerabilities with seismic fault lines. As long as the earth doesn’t move – you’re safe,
How to secure patient data in a healthcare organization
If you are a HIPAA covered entity or a business associate vendor to a HIPAA covered entity the question of HIPAA – the question of
Debugging security
There is an interesting analogy between between debugging software and debugging the security of your systems. As Brian W. Kernighan and Rob Pike wrote in
Encryption, a buzzword, not a silver bullet
Encryption, buzzword, not a silver bullet for protecting data on your servers. In order to determine how encryption fits into server data protection, consider 4
Treat passwords like cash
How much personal technology do you carry around when you travel? Do you use one of those carry-on bags with your notebook computer on top
Tahrir square – the high-tech version
From Wired The revolt that started a year ago today in Egypt was spread by Twitter and YouTube, or so the popular conception goes. But a group of Navy-backed
The megaupload bust
My daughter was distressed yesterday after the Feds shutdown the megaupload file sharing site – “How am I going to see all those series and
Clinical trials in the cloud
Ben Baumann from Akaza and Open Clinica fame, recently blogged about clinical trials in the cloud. Ben is pitching the relatively new offering from Akaza
Anatonme – a hand held device for improving patient-doctor communications
From a recent article in Healthcare Global. Studies suggest that 30-50 percent of patients are likely to give up treatments early. Microsoft Research has developed
Beyond the firewall
Beyond the firewall – data loss prevention What a simple idea. It doesn’t matter how they break into your network or servers – if attackers
Why web application security is fundamentally broken
Web application security in the cloud View more presentations from Software Associates
Are passwords dead?
A recent article on CSO online ponders the question of whether or not passwords are dead – since they are not much of a security
Data Classification and Controls Policy for PCI DSS
Do you run an e-commerce site? Are you sure you do not store any payment card data or PII (personally identifiable information) in some MySQL
